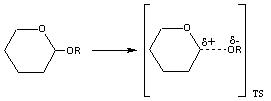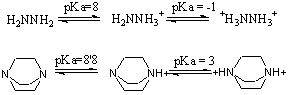|
Biopolímeros (4831) |
 |
|
Módulo 4. Ficha
4.7
|
|
|
Biopolímeros (4831) |
|
|
Módulo 4. Ficha
4.7
|
ENZIMAS
4.3. Conceptos de catálisis química.
4.3.4. Ejemplos de tipos de catálisis. Aplicación a la catálisis enzimática
4.3.4.1. Catálisis ácida y básica general
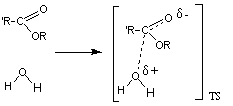
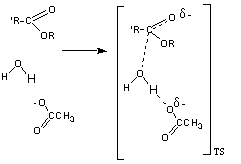
La reacción de hidrólisis no catalizada de un ester, implica el ataque nucleófilo de la molécula de agua neutra sobre el carbono carbonílico del ester, también neutro, para dar el estado de transición en el que una cierta carga positiva se desarrolla sobre el agua atacante y una cierta carga negativa sobre el oxígeno carbonílico
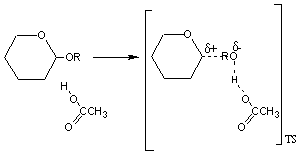
En la hidrólisis no catalizada de un acetal se desarrolla un estado de transición con una estructura próxima a un carbocatión y a un ión alcóxido En ambos casos las reacciones suceden a través de un estado de transición muy desfavorable debido a la separación de cargas. Desde luego, la estabilización de estas cargas, dará lugar a una disminución de la energía de activación y como consecuencia a un aumento en la velocidad de reacción.
La estabilización de la carga positiva desarrollada sobre el agua puede realizarse a través de la transferencia de uno de sus protones a una base. Este es un proceso de catálisis general básica El estado de transición puede estabilizarse por la transferencia de un protón desde un ácido al oxígeno del alcóxido que va a ser expulsado en la reacción. Este proceso se conoce como catálisis general ácida.
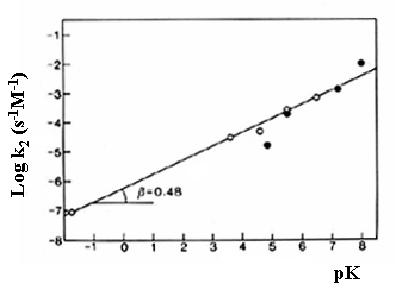
La existencia de catálisis general ácida o básica general puede ponerse de manifiesto realizando medidas de constantes de velocidad de la reacción a diferentes concentraciones totales del ácido o de la base, aunque manteniendo pH constante con el fin de no variar la concentración total de protones o hidroxilos y por tanto no alterar la posible existencia de catálisis ácida o básica específica. La representación de la constante de velocidad frente a la concentración de ácido o de base debe ser una línea recta, cuya pendiente es la constante específica de catálisis, k2. La variación del logaritmo de la constante específica de catálisis ácida o básica con el pKa de los catalizadores es una línea recta y constituye un ejemplo de las LFER de las que antes se habló. La eficacia de una catálisis general ácida o básica viene dada por la ecuación de Brönsted-Pedersen con pendiente positiva en el caso de la catálisis básica y negativa en el caso de la catálisis ácida:
log k2 = A + b pK
log k2 = A - a pK
Catálisis general básica para la hidrólisis del dicloroacetato de etilo. Los puntos vacíos, de oxianiones, y los puntos rellenos de aminas caen en la misma recta, lo que indica que la catálisis depende de la fortaleza de la base y no de su naturaleza química. Los valores de b y a varían entre 0 y 1, según que no haya catálisis o que la transferencia del protón sea total.
En catálisis enzimática, los procesos de catálisis ácida o básica general son muy frecuentes. Entre los aminoácidos esenciales, existen tanto grupos ácidos como básicos, que pueden ejercer esta función, no obstante por su valor de pK en torno a la neutralidad, a la cual suceden la mayoría de los procesoso biológicos, el grupo imidazol de la Histidina es uno de los más habituales catalizadores ácido-base general en la catálisis biológica.
4.3.4.2.Catálisis electrostática
Desde luego, en los ejemplos de TS presentados en la sección anterior, las cargas positivas y negativas pueden ser estabilizadas por la presencia de iones del signo contrario en un proceso que se denomina catálisis electrostática. La carga positiva del carbocatión del estado de transición se podría haber estabilizado por interacción con el campo eléctrico negativo de, por ejemplo, un carboxilato y la carga negativa desarrollada sobre el oxianión se puede estabilizar mediante la carga positiva de un metal como el Zn2+ o el Mg 2+.
La intensidad de las interacciones iónicas está fuertemente modulada por el disolvente (constante dieléctrica), el cual puede modular las interacciones entre iones cercanos, aunque realmente sus moléculas no se interpongan entre ellos. Este hecho puede ponerse claramente de manifiesto si observan los valores de pK de compuestos que dan lugar a especies di o triiónicas con el mismo signo de la carga. Por ejemplo los pK de ionización de diaminas
La ionización de la hidrazina da lugar a un compuesto con dos cargas positivas separadas una distancia de 1’5 Å. En el vacío esto daría lugar, por las interacciones electrostáticas surgidas, a una desestabilización de la molécula de unos 920 kJ/mol. Sin embargo la diferencia de pK es sólo de 9 unidades, lo que refleja una diferencia de energía de sólo 50’2 kJ/mol. Una cosa similar ocurre en el caso de la diamina cíclica, en donde la ionización de los dos grupos de la trietilendiamina produce un dicatión con una separación de cargas de 2’6 Å, lo que en el vacío debería producir una desestabilización de 546 kJ/mol, aunque la diferencia de sus pK indica una perturbación de sólo 33’4 kJ/mol. La estimación de la constante dieléctrica entre los dos nitrógenos, tanto en un caso como en el otro, da un valor de 17, el cual no se corresponde con el valor de la constante dieléctrica del agua (78). Esto es debida a que el agua, en este caso, no se está comportando como un dieléctrico en el sentido clásico de "moléculas de dieléctrico entre los iones", sino que los iones positivamente cargados están polarizando las moléculas del disolvente situadas a su alrededor y alterando por tanto el dipolo permanente del agua (de las moléculas de agua cercanas). Los campos electrostáticos producidos por estos dipolos, así como por la presencia de algunos de los contraiones, neutralizan parcialmente los campos positivos de los cationes, el resultado es un apantallamiento que equivale a una constante dieléctrica de 17. Así que, cuando los iones están rodeados por un dieléctrico, aun cuando sus moléculas no se interpongan entre los iones, se rebaja la energía de interacción entre ellos. Esta atenuación de los campos electrostáticos entre iones cuando las moléculas están en el seno del disolvente acuoso, es una de las causas por las que los fenómenos de catálisis electrostática en disolución acuosa son poco importantes.
Sin embargo, si la reacción de catálisis ocurre en el seno de una enzima las cosas varían tremendamente ya que la proteína es un medio heterogéneo en lo referente a constante dieléctrica. En muchos casos, en el sitio activo, en donde tiene lugar la reacción catalítica, el agua está prácticamente excluida y en un medio apolar las interacciones electrostáticas son mucho más fuertes que en agua, debido a la baja constante dieléctrica. Los cálculos realizados sobre estabilización de cargas en entornos enzimáticos, teniendo en cuenta las constantes dieléctricas locales y su efecto sobre dipolos locales permanentes o inducidos permiten concluir que, en general:
- Dos o tres dipolos fijos en una enzima (por ejemplo los grupos =NH del esqueleto) pueden estabilizar una carga tan eficazmente como si estuviera en agua pura.
- Los dipolos fijos de un enzima pueden estabilizar un par iónico mejor que el agua pura
La justificación de estos dos hechos hay que encontrarla en la manera en la que se produce la solvatación de iones en el agua. En primer lugar, el agua forma una primera esfera de solvatación muy tensionada alrededor del ión. Rodeando a esta primera esfera se forman otras esferas de solvatación por la interacción de los dipolos electrostáticos del agua. Las moléculas de estas esferas externas de solvatación interacciónan también con las moléculas del bloque del disolvente mediante enlaces de hidrógeno e interacciones dipolares y como consecuencia, las interacciones de la capa externa de solvatación se debilitan y pierden una orientación definida. Los cálculos realizados, indican que la energía de solvatación mayor se alcanza cuando el ión está rodeado por unas diez moléculas de agua y no, cuando se considera todo el disolvente en bloque. Las enzimas, como puede verse en la figura siempre utilizan partes de su propia estructura o de iones unidas a ellas para solvatar los estados de transición, es decir, no utilizan el disolvente (en bloque) para la estabilización de estos estados.
Ficha
anterior 
|
 Ficha
Siquiente Ficha
Siquiente |
 |