 |
Biopolímeros (4831) |
 |
|
Módulo 2. Ficha
2.8
|
|
|
Biopolímeros (4831) |
|
|
Módulo 2. Ficha
2.8
|
FUERZAS QUE DETERMINAN LA ESTRUCTURA MACROMOLECULAR
2.8.Las interacciones hidrófobas
La fuerte inclinación de las moléculas de agua a formar enlaces de hidrógeno con otras moléculas, determina su comportamiento frente a moléculas no polares, incapaces de formar enlaces de hidrógeno, tales como alcanos, hidrocarburos en general , fluorcarburos, átomos inertes. Cuando las moléculas de agua entran en contacto con este tipo de moléculas se encuentran con un aparente dilema: cualquiera que sea el sitio al que la molécula de agua enfoque, parece que una o más de las cuatro cargas moleculares tiene que apuntar hacia la molécula inerte de soluto y por tanto pierde capacidad de formar enlaces de hidrógeno. Claramente la mejor configuración sería la de tener el menor número de cargas tetraédricas apuntando hacia la especie de soluto, de modo que las otras cargas pudieran apuntar hacia la fase acuosa siendo por tanto capaces de participar en enlaces de hidrógeno, tanto como antes.
Hay varias disposiciones estructurales que el agua puede adoptar para salvar los enlaces de hidrógeno perdidos. Si la molécula de soluto no es demasiado grande, es posible para las moléculas de agua empaquetarse alrededor de ella sin perder ninguno de sus sitios de enlace de hidrógeno formando asociaciones tridimensionales como la que se muestra en la figura de más abajo. Si las moléculas son mayores, los clusters formados pueden adaptarse a la forma y tamaño de la molécula de soluto.
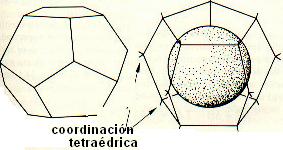 |
Formación de clusters entre las moléculas de agua, con coordinación tetraédrica y alojamiento de una molécula no formadora de enlaces de hidrógeno en su seno. |
Por tanto, el dilema anteriormente mencionado, tiene muchas veces una solución fácil, gracias a la posibilidad de unión de las moléculas de agua tetraédricamente coordinadas alrededor de casi cualquier molécula inerte, cualquiera que sea su tamaño o form. Del dibujo de la figura también se deduce que el agua que está alrededor de la molécula de soluto inerte tiene una mayor coordinación (cuatro) que las moléculas de agua en el líquido puro (3'0-3'5 en promedio) y por tanto que esta disposción de clusters está incluso favorecido desde el punto de vista energético. A este fenómero se le conoce con el nombre de solvatación hidrófoba o hidratación hidrófoba. Es conocido que a grandes presiones los hidrocarburos y los átomos inertes pueden disolverse en agua. Los calores de hidratación (solvatación hidrófoba) medidos para estos compuestos pueden verse en la siguiente tabla (Datos del libro de Cantor and Schimel)
|
Calores de hidratación de
sustancias apolares
|
|
|
Molécula |
- DH(kcal/mol) |
|
Ar |
16'6 |
|
Kr |
13'9-16'5 |
|
CH4 |
14'5-17 |
|
C2H2 |
15 |
|
C2H4 |
15 |
|
C2H6 |
15 |
|
CH3SH |
16'6 |
Como puede observarse, las reacciones son exotérmicas y todas ellas tienen, prácticamente, la misma energía; lo que confirma lo anteriormente expuesto, en el sentido de que este balance energético no hay que achacarlo a interacciones soluto-disolvente, sino a reorganización de las interacciones en el disolvente. Esta reorganización de las moléculas de disolvente, aunque sea energéticamente favorable, es entrópicamente desfavorable ya que rompe la anterior estructura del agua líquida e impone en las moléculas de agua que rodean al soluto una nueva y más ordenada y por tanto con menor entropía.
Se han calculado el valor de diferentes funciones termodinámicas de transferencia de hidrocarburos, es decir las funciones termodinámicas ligadas al proceso de tomar una molécula de hidrocarburo que está en el hidrocarburo líquido puro e introducirla en agua pura a la misma temperatura. La siguiente tabla ilustra el valor de estas funciones en donde se pone claramente de manifiesto que el motivo de que la disolución de hidrocarburos en agua no sea un proceso favorable es de naturaleza entrópica.
| Parámetros termodinámicos de transferencia de hidrocarburos desde el disovente apolar a agua a 25º | |||
|
Reacción de transferencia |
DH (kcal/mol) |
DS (cal/mol K) |
DG (kcal/mol) |
|
CH4 en benceno -> CH4 en agua |
-2'8 |
-18 |
+2'6 |
|
CH4 en eter -> CH4 en agua |
-2'4 |
-19 |
+3'3 |
|
CH4 en CCl4 -> CH4 en agua |
-2'5 |
-18 |
+3'3 |
|
C2H6 en benceno -> C2H6 en agua |
-2'2 |
-20 |
+3'8 |
|
C2H6 en CCl4 -> C2H6 en agua |
-1'7 |
-18 |
+3'7 |
|
C3H8 líquido -> C3H8 en agua |
-1'8 |
-23 |
+5'5 |
|
n-C4H10 líquido -> n-C4H10 en agua |
-1'0 |
-23 |
+5'85 |
Es interesante observar que, para los diferentes hidrocarburos, la energía libre de transferencia es aproximadamente proporcional a área superficial de las moléculas, lo que es un claro indicativo de que el número de moléculas de agua reorientadas está más o menos determinada por las áreas en las que no hay enlaces de hidrógeno. Así para el metano cuyo radio de Van der Waals a=s/2 = 0'2 nm, el área superficial por molécula vale 4pa2 » 0'50 nm2 y la energía libre de transferencia (DG = 13'8 kJ/mol) por unida de área superficial vale alrededor de 46 mJ/m2. Similarmente, para una molécula de n-butano cilíndrica, cuya superficie viene dado por 4pa2 + 2pa (3x0'1275) » 1'0 nm 2 y teniendo en cuenta el valor de la energía de Gibbs para este compuesto, se deduce una energía por unidad de área de 41 mJ/m 2. Estos valores son muy similares al valor de la energía libre interfacial (g i) de la interfacies hidrocarburo-agua que generalmente tiene valores entre 40 y 50 mJ/m 2.
La inmiscibilidad de sustancias inertes con agua y la naturaleza fundamentalmente entrópica de esta incompatibilidad se conoce como el efecto hidrófobo y a estas sustancias, sustancias hidrófobas. Directamente relacionado con el efecto hidrófobo está la interacción hidrófoba que describe las inusualmente fuertes interacciones entre estas moléculas cuando están en un entorno acuoso. Por ejemplo, las interacciones de van der Waals entre dos moléculas de metano en contacto en el espacio libre es -2'5. 10 -21 J, mientras que en agua es - 14. 10-21 J. De manera similar, las tensiones superficiales de la mayoría de los hidrocarburos cae entre 15-30 mJ/m2, mientras que sus tensiones interfaciales con agua están en el rango de 40-50 mJ/m2.
Debido a su fortaleza se creyó originariamente que el responsable de esta interacción era algún tipo de "enlace hidrófobo . Pero debe quedar claro que no hay enlace asociado con este fenómeno, el cual tiene causas fundamentalmente entrópicas que proceden principalmente de la reordenación de la configuración de los enlaces de hidrógeno en las regiones solapantes de solvatación de dos especies hidrófobas cuando están próximas y por tanto de mucho más largo alcance que cualquier enlace típico.
Las figuras siguientes ilustran el hecho de la formación de interacciones hidrófobas teniendo en cuenta el balance del coste entrópico que supone mantener el sistema (a) en lugar del (b). Verdaderamente en la situació (b) se forman menos enlaces de hidrógeno que en la (a), pero este inconveniente energético se compensa con un aumento de las interacciones de van der Waals entre los solutos y sobre todo con el balance favorable entrópico que supone adquirir aquella distribución molecular.
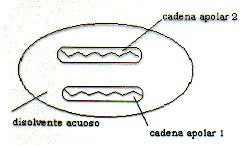 (a) (a) |
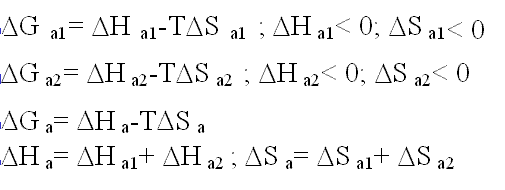 |
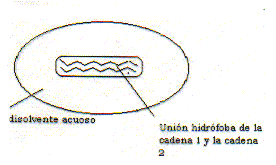 |
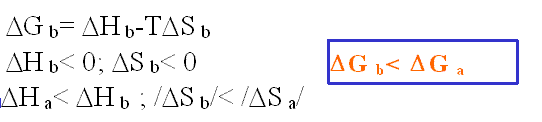 |
¿En cuanto se puede estimar el valor de la interacción hidrófoba? Hay pocas medidas directas de las interacciones hidrófobas entre moléculas no polares disueltas, principalmente porque son insolubles en condiciones moderadas de presión y temperatura. Tucker y colaboradores han dado valores entre -8'4 y -11'3 kJ/mol para las energías de dimerización benceno-benceno y ciclohexano-ciclohexanol respectivamente y Ben Naim y colaboradores dedujeron un valor de aproximadamente -8'6 kJ/mol para dos moléculas de metano.
Teóricamente el problema no es fácil de abordar, debido a que la interacción hidrófoba entre dos moléculas es muy compleja, implica a muchas otras moléculas y es de mayor alcance que cualquiera de los potenciales entre pares de moléculas sencillos estudiados hasta ahora. Israekachvili y Pashley en 1982 midieron la fuerza hidrófoba entre dos superficies hidrófobas macroscopicamente curvadas en agua y encontraron que en el rango de 0 -10 nm las fuerzas caían exponenciamente con la distancia. Basándose en estos experimentos, estos autores propusieron para las pequeñas moléculas de soluto que la energía libre de dimerización hidrófoba aumenta proporcionalmente con su diámetro de la forma:
DG (potencial del par hidrófobo) »-20 s kJ/mol
donde s está en nanometros. Por ejemplo para el ciclohexano (s = 0'57 nm) esta energía es DG » -11'4 kJ/mol en concordancia con el valor medido.
Las interacciones hidrófobas juegan un papel central en muchos fenómenos de superficie y de autoasociacion molecular, en la formación de micelas, en la estructura de las membranas biológicas, en la determinación de las conformaciones de las proteínas, en los efectos de stacking entre los anillos aromáticos de las bases púricas y pirimidínicas que forman las cadenas de ácidos nucleicos, etc.
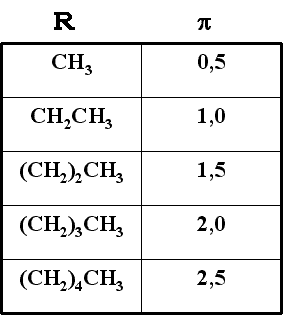
Existen diversas escalas para medir la hidrofobicidad de los grupos químicos. Una de ellas utiliza el parámetro de hidrofobicidad p . Para un grupo químico R, p viene definido por la expresión:
p = log (S/So)
En donde S es la relación de solubilidad molar de A-R en octanol y agua y So es la relación de solubilidad molar de A-H en octanol y agua. En esta escala los valores positivos de p indican grupos hidrófobos y los valores negativos indican grupos hidrófilos. Aunque el coeficiente hidrófobo total no es igual, en la mayoría de los casos, a la suma de los coeficientes de los diferentes grupos, en el caso de algunos compuestos como los hidrocarburos, en donde existe una gran similitud estructural de los diferentes grupos, sí que se da esa aditividad como se puede apreciar en los datos de la tabla siguiente.
Ficha
anterior 
|
 |