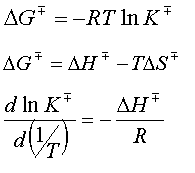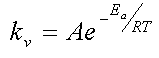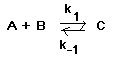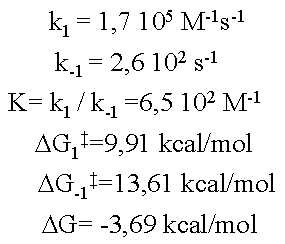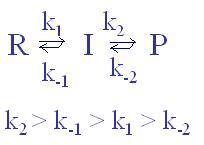|
Biopolímeros (4831) |
 |
|
Módulo 4. Ficha
4.5
|
|
|
Biopolímeros (4831) |
|
|
Módulo 4. Ficha
4.5
|
ENZIMAS
4.3. Conceptos de catálisis química.
4.3.2. La Teoría del Estado de Transición (TET).
4.3.2.1. Las Funciones Termodinámicas de Activación: Energía libre de Gibbs, Entalpía y Entropía de Activación
La constante de equilibrio, K‡ , de la ecuación anterior puede relacionarse con la variación de energía libre de Gibbs, DG‡ ,entre los dos estados o Energía Libre de Gibbs de Activación.
que es la diferencia de energía libre de Gibbs entre el Complejo Activado y los reactivos. Recuerdese que el Complejo Activado es una molécula, formada por todos los átomos que forman los reactivos, aunque con un grado de libertad vibracional menos (la coordenada de reacción).
De igual forma y teniendo en cuenta las relaciones entre las funciones termodinámicas, pueden introducirse los conceptos de Entalpía de Activación, DH‡,como la diferencia entre la entalpía del Complejo Activado y los reactivos y la Entropía de Activación, DS‡, como la diferencia entre la entalpía del Complejo Activado y los reactivos.
La entalpía de activación no es la Energía de Activación, Ea, de la Ecuación de Arrhenius:
pero está relacionada directamente con ella. Realmente, los parámetros de la Ecuación de Arrhenius, Ea y A se relacionan con las funciones termodinámicas del proceso de activación según las siguientes ecuaciones:
La relación entre la constante de velocidad y las energías de Gibbs de los estados, permite representar las transformaciones a lo largo de la coordenada de reacción en términos de variaciones de energía libre y obtener diagramas de la coordenada de reacción análogos a los obtenidos sobre la SEP, aunque hay que tener en cuenta, que la energía potencial es un concepto mecánico aplicable a una partícula individual, mientras que la energía libre es un concepto termodinámico aplicable a un conjunto grande de partículas.
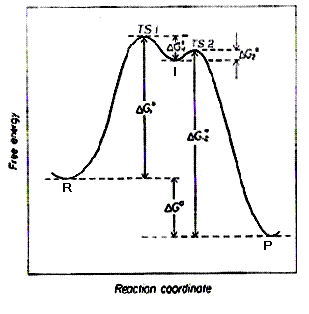
El complejo activado no es un intermedio de reacción. En la figura se respresenta un diagrama de coordenada de reacción con un intermedio (I, un mínimo de energía) y dos complejos activados, TS1 y TS2. La transformación global será:
4.3.2.2. El postulado de Hammond
En la figura anterior el estado de transición se ha situado más cerca de los reactivos que de los productos en aplicación del postulado de Hammond que establece que el TS de una reacción elemental está más cercano a aquel estado (inicial o final) que tiene la mayor energía. Desde luego el postulado puede justificarse por un lado, en el hecho de que si dos especies que están relacionadas por una transformación química tienen energías similares, probablemente también tendrán estructuras similares (fundamento químico) y por otro, en el hecho de que si dos estados que se dan consecutivamente sobre la coordenada de reacción tienen energías similares, su interconversión requerirá solo pequeños cambios en sus estructuras.
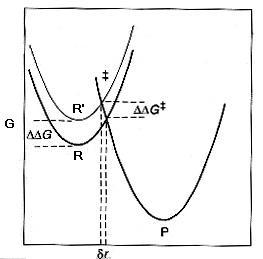
En la figura se representa esquemáticamente el punto de intersección entre las curvas vibracionales del reactivo R y el producto P en donde se situa el TS. Si se hace una pequeña modificación estructural en el reactivo, de forma que el nuevo reactivo R' esté algo desestabilizado con respecto a la primera situación (DDG), el nuevo punto de corte estará desplazado (dr) hacia la posición del estado que se ha desestabilizado. El nuevo TS' estará desestabilizado DDG‡ respecto del inicial La figura anterior explica la base de lo que se conoce con el nombre de relaciones lineales de energía libre, LFER, (linear free energy relationships), es decir cambios en las energías libres del equilibrio (DDG) están directamente relaciondas con cambios en las energías libres de activación (DDG‡ ) y por tanto, los logaritmos de las constantes de equilibrio se relacionan de forma lineal con los logaritmos de las constantes cinéticas. Este movimiento a lo largo de la coordenada de reacción se conoce con el nombre de el efecto Hammond.
Ficha
anterior 
|
 Ficha
Siquiente Ficha
Siquiente |
 |